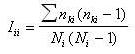|
Autor: Índice general Introducción La primera constancia
que se tiene del uso de los apellidos con fines genéticos
es la efectuada por G. H. Darwin en 1875, merced a la cual éste
logró estimar la frecuencia de matrimonios consanguíneos
entre primos hermanos en población inglesa. Aunque inicialmente
el término Isonimia fue concebido como la propiedad
inherente a aquellos matrimonios, cuyos cónyuges poseían
el mismo apellido, pronto quedó establecido como la metodología
consistente en el uso de los apellidos presentes en una determinada
población, para el estudio de la estructura genética
de la misma [1]. Mediante el empleo de los apellidos
es posible estimar numerosos parámetros relacionadas con
la genética de la población, entre los cuales figuran
los estadísticos F, consanguinidad, parentesco interpoblacional,
etc. Ello es posible, dado que los apellidos se transmiten transgeneracionalmente,
de padres a hijos, siguiendo un patrón de transmisión
que guarda relación directa con la herencia genética. El concepto
de Isonimia, como método de estudio en la Genética
de Poblaciones Humanas, nace en el año 1965, en virtud del
trabajo presentado por Crow y Mange [1]. Inicialmente
el campo de aplicación de la isonimia se limitaba al cálculo
de la consanguinidad, aunque ya en 1968, Lasker utilizó listas
de apellidos para la estimación del parentesco genético
entre poblaciones humanas; pese a lo cual no logró una formulación
adecuada a este fin hasta el año 1977 [2].
Formulaciones de métodos análogos para la estimación
del parentesco genético entre poblaciones, han venido siendo
realizadas introduciéndose puntualizaciones metodológicas,
cada vez más precisas para su estimación contribuyendo
de forma decisiva al perfeccionamiento de la Isonimia [3].
El desarrollo e instauración de estas técnicas se
ha visto arropado por el notable avance registrado en material y
procedimientos informáticos, lo cual ha contribuido a la
proliferación de trabajos, cuyo substrato metodológico
es la Isonimia, pese a lo cual en el País Vasco únicamente
destacan los trabajos realizados por Peña [4],
Delgado [5] y Rodríguez, Delgado e Izarzugaza
[6, 7]. 2. Elaboración de matrices de parentesco genético. Una de las aplicaciones más importantes de la isonimia, es la elaboración de matrices de parentesco genético que relacionen diferentes poblaciones entre sí, al tiempo que permiten calcular diferentes estadísticos de consanguinidad (F). El parentesco genético es la probabilidad de que tomando un gen de una población i, éste sea idéntico por descendencia a otro gen localizado en la población j, surge el parámetro sij, que cuantifica dicho parentesco. El método de Morton [3], permite el cálculo de una estimación de dicho parámetro mediante isonimia. Dicho método parte del concepto de isonimia esperada, dicha isonimia esperada puede ser calculada a distintos niveles poblacionales: intrapoblacional, entre pares de poblaciones y a nivel regional. La isonimia esperada para una población (i), queda expresada mediante la siguiente ecuación:
(I) 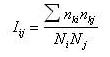 (II) Finalmente se puede calcular la isonimia a nivel regional (R), considerando el número total de las poblaciones que componen el área de estudio de modo simultáneo, o lo que es lo mismo la totalidad de subpoblaciones (p) que componen la población total: 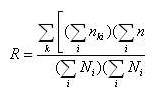 (III) Una vez introducido el concepto de isonimia esperada a sus distintos niveles: poblacional, pares de poblaciones y regional; nos hallamos en condiciones de exponer los tres coeficientes de parentesco diseñados por Morton: parentesco a priori, parentesco regional y parentesco condicional. La totalidad de dichos coeficientes de parentesco son conceptualmente distintos, calculándose de forma diferente en cada caso, aunque todos ellos permiten elaborar una matriz de parentesco de dimensiones pxp, siendo p el número de poblaciones que componen el estudio o si se prefiere el número de subdivisiones o subpoblaciones en que se fragmenta la población general, conforme a alguno de los criterios antes mencionados. El parentesco a priori se apoya conceptualmente en la presunta existencia de una población primigenia, la cual pudo haber dado origen a las poblaciones actuales o contemporáneas [3]. La existencia de dicha población hipotética ha de situarse en el pasado y su constatación es puramente especulativa. El parentesco a priori es matemáticamente un cuarto de la isonimia esperada entre pares de poblaciones, descrita anteriormente: 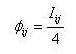 (IV) Otro coeficiente de parentesco, es el denominado coeficiente de parentesco regional, el cual se define como la cuarta parte de la isonimia esperada a nivel regional: (V) No obstante el coeficiente de parentesco más usual y empleado con mayor frecuencia en los estudios basados en estructura genética de poblaciones humanas, es el denominado coeficiente de parentesco condicional [3, 5]. Dicho coeficiente se define como un coeficiente de covarianzas de las frecuencias génicas de las poblaciones en torno a una media regional, dicho de otro modo, se trata de un coeficiente construido en base a covarianzas subpoblacionales en torno a la media de la población general en su conjunto [3, 5]. Matemáticamente queda expresado mediante la siguiente ecuación: 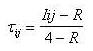 (VI) Finalmente se ha de constatar la relación existente entre los dos coeficientes de parentesco: condicional y a priori, mediante las siguientes expresiones: 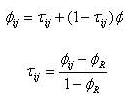 (VII) (VIII) 3. Utilidad del estudio de la Isonimia en Medicina y Epidemiología. Hasta el momento
el estudio de la asociación entre diferentes genes y el desarrollo
de cáncer de diferentes localizaciones, se basa en estudios
costosísimos de ligamiento genético. Por ejemplo,
el cáncer de mama constituye una de las principales causas
de muerte entre la población femenina mundial, al tiempo
que es el más frecuente entre los diferentes cánceres
susceptibles de afectar dicha población. Recientes descubrimientos
en el ámbito de la Epidemiología Genética han
demostrado la existencia de oncogenes asociados a la aparición
de la enfermedad, entre los cuales destacan diferentes alelos pertenecientes
a los loci BRCA1 y BRCA2 [6, 7].
Estos genes de susceptibilidad al cáncer de mama aparecen
ligados también a cáncer de ovario, colon, páncreas
y próstata. No obstante, estos genes únicamente explican
una determinada proporción de los casos registrados, según
investigaciones recientes, el resto de casos podrían ser
debidos a otros genes, cuya investigación se encuentra en
curso, o bien deberse a causas exclusivamente medioambientales.
Pese a la sencillez del método genético de análisis de Isonimia, se plantean numerosos problemas informáticos a la hora de introducir y manejar los datos por su elevado volumen y la gran cantidad y complejidad de operaciones matemáticas a efectuar. El objetivo fundamental es la construcción de diferentes rutinas, basadas en un programa de análisis estadístico, capaz de tratar un volumen importante de datos y realizar otro tipo de análisis con los resultados obtenidos. En un primer momento las rutinas se desarrollaran para el paquete estadístico STATA® [8] pero pueden ser implementadas en otros paquetes como SAS o SPSS. Mediante STATA®,
se elaboraron una serie de rutinas informáticas encadenadas,
las cuales permiten la estimación del parentesco genético
en sus dos vertientes: parentesco a priori y parentesco condicional.
A su vez permite la elaboración de matrices de parentesco
genético y distancias genéticas, las cuales pueden
ser utilizadas en numerosos contextos metodológicos, genéticos,
antropológicos y médico- Describimos a continuación una serie de programas que nos permiten la estimación mediante isonimia de las matrices de parentesco y distancia genética entre diferentes subpoblaciones pertenecientes a una misma población objetivo de interés para nuestro estudio. Los programas deben ejecutarse en el orden en que los describimos en este apartado, ya que los resultados del primero son utilizados por los siguientes. Al final de este trabajo, se recogen en un anexo, el listado completo de las instrucciones utilizadas en el lenguaje de STATA 7.0. a) Preparación de los datos para la estimación de isonimia. El proceso informático comienza con la preparación de los datos para la posterior estimación de los coeficientes de Isonimia. El programa supone que se parte de un fichero de datos cargado en la memoria de STATA que consta de cierto número de casos o individuos de la población de interés (filas), en los que se han medido diferentes variables (columnas). El número máximo de sujetos que pueden ser incluidos dentro del análisis, viene limitado únicamente por la memoria RAM de la máquina donde se ejecuta el programa. Cada individuo es identificado mediante una única variable (obse) durante todo el resto del proceso informático. Entre las variables deben figurar una o dos de tipo cadena alfanumérica, que recojan el primer y segundo apellido de cada individuo. No es obligatorio que existan dos apellidos, el programa funciona también con un único apellido, como ocurre en poblaciones de origen anglosajón. El resultado
de este programa es un fichero de datos en el que cada fila se corresponde
con un apellido del total de los apellidos considerados en la población
analizada. De manera que si cada individuo tiene dos apellidos,
el fichero de salida contiene el doble de filas que los datos originales.
Se graban también dos ficheros separados, que contienen la
lista de apellidos paternos y maternos, si estos últimos
existen en el fichero original. El segundo programa convierte los apellidos, originalmente cadenas alfanuméricas, en datos numéricos. A cada apellido se le asigna un código numérico, con lo que durante todo el proceso se preserva la identidad de los sujetos objeto de estudio, al tiempo que se racionaliza el uso de memoria y recursos de la máquina donde se ejecuta el programa. El resto de las variables medidas sobre cada individuo se conservan intactas. La rutina proporciona
diferentes salidas que son guardadas en la memoria o en diferentes
ficheros. En primer lugar, se calcula el número total de
apellidos en la población de interés (no missing)
y el número de los apellidos que son diferentes entre sí.
En segundo lugar, se salva un fichero conteniendo la equivalencia
numérica de todos los apellidos, que puede ser utilizado
posteriormente para reconstruir los datos originales (codeape.do).
Por último, el programa guarda las frecuencias absolutas
de cada uno de los apellidos de la población de interés
en un fichero de datos (apefreq.dat). Este programa permite al investigador establecer las subpoblaciones a considerar a partir de combinaciones de valores de las variables seleccionadas por el usuario. Dichas variables deben haber sido medidas sobre los individuos y recogidas en el fichero de datos original sobre el que se inició el análisis. La definición de poblaciones puede incluir variables continuas, pero esto conduciría a establecer un número muy alto de subpoblaciones, por lo que desaconsejamos la utilización de este tipo de variables en la definición de subpoblaciones. En cualquier caso, el programa informa del número de subpoblaciones definidas (p). El número máximo de poblaciones que se pueden definir, viene limitado por la capacidad de STATA® para trabajar con matrices dependiendo de la versión de STATA®: 40 poblaciones en la versión standard y 800 en Intercooled STATA®. Con STATA® SE (special edition) el número de poblaciones sólo tiene el límite de la memoria física RAM instalada en el computador. Siguiendo la metodología descrita por Morton y Relethford [3] se genera la matriz de denominadores de cada isonimia esperada tanto intra como inter-subpoblacional. La matriz es cuadrada de dimensiones pxp (siendo p el número de subpoblaciones diferentes definidas previamente). Su diagonal contiene los valores Ni*(Ni-1), para i=1, 2,..., p. Es decir, los denominadores de la isonimia esperada dentro de cada subpoblación i (Iii). Los elementos fuera de la diagonal recogen los valores de los denominadores de la isonimia esperada entre cada par de subpoblaciones (Iij), es decir, Ni*Nj, para todo j diferente de i. Dichos denominadores, son los recogidos en las ecuaciones I y II ya mencionadas previamente. Además
de la matriz de denominadores guardada en un macro de memoria (Denom_isonymy),
el programa salva un fichero con todas las subpoblaciones definidas
(poblas.dta) y añade el código de población
a cada uno de los registros de apellidos incluidos en el análisis. El programa
calcula el estimador de la Isonimia a nivel regional, partiendo
de una única variable o argumento del programa, es decir
los códigos numéricos de los apellidos en el fichero
de datos de la población de interés. La salida se
almacena en ciertas variables o macros de memoria: el numerador
(R_numerator) y denominador (R_denominator) de la Isonimia regional
(R) mencionada en la ecuación III. Por último, el
cálculo del parentesco regional (R_kindship) es simplemente
un cuarto de la isonimia regional ya calculada. El programa sólo necesita dos argumentos que son el nombre de las dos variables que contienen los códigos de subpoblación y apellido. Es necesario poner en primer lugar, la variable que contenga los códigos de las subpoblaciones definidas previamente. El bucle principal del programa se encarga de calcular la matriz de dimensiones pxp de los numeradores de la isonimia intra e inter-subpoblacional como se recogen en las ecuaciones I y II. Para construir la matriz de isonimia (Isonymy) sólo es necesario dividir cada elemento de la matriz de numeradores, entre el elemento correspondiente de la matriz de denominadores calculados en una rutina previa y que se mantienen en la memoria de STATA ® mientras no se cierre el programa. Una vez calculada
la matriz de isonimia, se realiza el cálculo de dos matrices
de distancias genéticas entre cada par de subpoblaciones.
Ambas matrices son de dimensiones pxp. La primera de dichas matrices
(Gen_dist1) sólo es la distancia cuadrática entre
subpoblaciones bajo ciertas circunstancias. La segunda matriz (Gen_dist2)
siempre es cuadrática y su cálculo se realiza mediante
la siguiente expresión: La matriz de parentesco a priori (Pri_parent) representado en la ecuación IV se calcula dividiendo cada elemento de la matriz de isonimia entre el escalar 4. El parentesco condicional se construye según la ecuación VI mediante operaciones sencillas sobre los elementos de la matriz de isonimia. Por último, se construye el vector de varianzas génicas FST a partir de la diagonal de la matriz de isonimia (isonimia intra-subpoblacional) y la frecuencia relativa o proporción de apellidos en cada subpoblación. La salida de
esta última rutina es una serie de archivos en los cuales
se incluyen separada y ordenadamente las matrices de: Isonimia,
matriz de parentesco a priori, matriz de parentesco condicional,
matrices de distancias genéticas, vector diagonal de matrices
de parentesco y valor de FST (varianza de las frecuencias génicas).
La validación del programa se realizó a dos niveles: informático y epidemiológico. En ambos se utilizaron los casos de cáncer de mama y otras localizaciones del Registro de Cáncer del País Vasco, que incluía los dos apellidos de todos los individuos diagnosticados de cáncer en la población residente en el País Vasco desde 1986 hasta 1993. El número de casos que utilizamos en la validación superaba ampliamente los 36.000 y pudimos analizarlos y compararlos con los resultados obtenidos mediante los programas del profesor Rodríguez Larralde en la Universidad de Caracas (Venezuela) y los obtenidos a partir de otro programa construido por diferentes profesores de la universidad de Coimbra (Portugal). Los resultados eran idénticos hasta donde pudimos comparar, debido a que los resultados de STATA permiten una mayor precisión (4 decimales más). Los resultados
epidemiológicos de comparación de las distancias genéticas
entre poblaciones con cáncer de diferentes localizaciones
morfológicas, se recogieron en dos comunicaciones presentadas
a sendos Congresos de la Sociedad Española de Epidemiología
en los años 2001 y 2002 y publicadas en la revista Gaceta
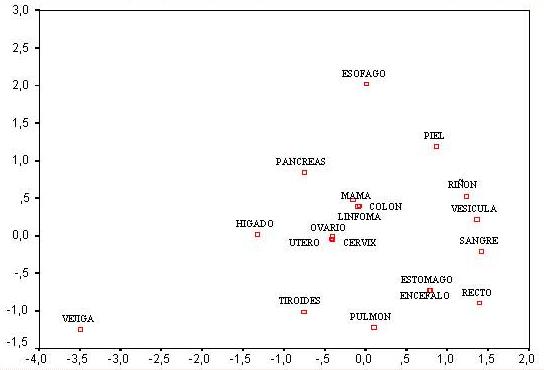
Sanitaria [6, 7]. En la Figura
1 se puede apreciar los resultados del análisis de la matriz
de distancias genéticas entre las subpoblaciones de cáncer
con diferente localización anatómica. Se puede apreciar
dos clusters o grupos de poblaciones más cercanas entre si.
El primer cluster corresponde a las que tienen localizado el cáncer
localizado en mama, útero, ovario, cervix y los que padecen
linfoma. El segundo cluster, está formado por colón,
ovario y recto. Lo que nos permitió concluir:
Agradecimiento
Bibliografia [2] Lasker GW. A coefficient of relationship by isonymy: A method for estimating the genetic relationship between populations. Hum. Biol. 1977; 52:311-324. [3] Relethford JH. Estimation of kinship and genetic distance from Surnames. Hum. Biol. 1988; 60: 475-492. [4] Peña JA. Estructura demográfica y genética de la población del Valle de Orozco (Vizcaya) Siglos XVI-XX. Tesis doctoral. Departamento de Biología Animal y Genética, Universidad del País Vasco (España). 1988. [5] Delgado Naranjo J. Estructura genética de la población de Lanciego, Mediante Isonimia (1800-1979). Tesis de licenciatura. Departamento de Biología Animal y Genética, Universidad del País Vasco (España). 1994. [6] Rodríguez Andrés C, Delgado Naranjo J. Análisis de la estructura genética mediante Isonimia de la población afectada de cáncer ligado a BRCA1, BRCA2 y otros genes de susceptibilidad al cáncer de mama. Gac Sanit 2001; 15 (Supl 2): 95-96. [7] Rodríguez Andrés C, Delgado Naranjo J, Izarzugaza Lizarraga I. Análisis de la estructura genética mediante Isonimia de la población de mujeres diagnosticadas cáncer ligado a BRCA1, BRCA2 y otros genes de susceptibilidad al cáncer de mama en el País Vasco. Gac Sanit 2002;16(Supl 1):118-119. [8]
STATA 7.0 User's Manual (2001). STATA Corporation. 4905 Lakeway
Drive. College Station TX77845 (USA). Anexo: Programas realizados en STATA® 7.0. a. Preparación de los datos para la estimación de isonimia. */ P19_1 Programa dosape program define dosape */ Crea la variable _obse.(ordenación segun primer apellido
y segundo apellido) if "`ape2'"!="" { b. Conversión de apellidos en indicadores numéricos. */ P19_2 Codificación Numérica de los apellidos. program define apeanum, eclass c. Define subpoblaciones y calcula la matriz de denominadores de la Isonimia. */ P19_3 program define npobla, eclass di in yellow "3. The number of Populations included in this
analysis is " _N
*/ P19_4_2 program define R_Isonymy, eclass
di in yellow "5. Numerator of (R) Regional Random Isonymy
Coeficient: " e(R_numerator) e. Matrices de Isonimia esperada, parentesco y distancia genética.
program define IsoNum, eclass
|