| |
Titulo: EL CÓDIGO GENÉTICO DESDE LA PERSPECTIVA
DE LA BIOINFORMÁTICA.
Autores:
Miguel Sautié Castellanos*, Carlos M. Martínez Ortiz*
, Osmel Companioni Nápoles** y José L Hernández
Cáceres*.
* CECAM, Centro de Cibernética Aplicada a la Medicina,
Instituto Superior de Ciencias Médicas de la Habana.
** CIGB, Centro de Ingeniería Genética y Biotecnología,
Departamento de Genómica.
Índice
general
Resumen
El Código Genético Standard (CGS) no es el resultado de un proceso de asignación aleatoria de aminoácidos a codones, sino más bien todo lo contrario, determinadas regularidades estructurales que lo distinguen y que tienen profundas consecuencias en las propiedades de las secuencias biológicas actuales y en sus patrones evolutivos, revelan el resultado de la acción de ciertas leyes. La bioinformática ha jugado un rol central no sólo en la evaluación de la repercusión de estas características del CGS sino también en la búsqueda de las causas, las fuerzas químico-fisicas, los mecanismos evolutivos que conformaron su estructura actual o los que dieron lugar a algunas de sus variantes nucleares u organulares.
En general el desarrollo de las informática han revolucionado la metodología de las ciencias empíricas, un nuevo empirismo in silico han permitido la implementación de modelos matemáticos y la ejecución de nuevos experimentos de validación con ventajas enormes en cuanto a costo y tiempo, la biología evolutiva no ha sido ajena a la influencia de estos cambios, en este trabajo se podrá apreciar la decisiva participación de diferentes métodos computacionales en la aclaración aún incompleta de algunas incógnitas relacionadas con el CGS.
Introducción
El código genético es un conjunto de reglas de asignación de aminoácidos a tripletes de bases nitrogenadas, en este se define el vocabulario y los principios de conversión del genotipo en fenotipo por medio de sus unidades más elementales, el código representa uno de los cambios evolutivos más importantes, su aparición fue resultado de la división fundamental de la función catalítica (ARN y polipéptidos) con respecto a la informacional (ARN) ocurrida posiblemente en antiguos progenotes ribonucleopeptídicos progenitores del llamado Ultimo Ancestro Común Universal (LUCAS). De ahí que casi todos los organismos conocidos y pertenecientes a los tres reinos Archea, Eucaria y Procariota tengan una biología regida por estos principios "prima facie" sólo en algunos de ellos estos se presentan con algunas modificaciones que se repiten en muchos casos en organismos provenientes de reinos diferentes. Casi desde que se descifró el código genético canónico en la década del 60 muchos observaron que la organización de las reglas de asignación no era aleatoria. En este se pueden detectar fácilmente patrones tanto "horizontales" en la relación entre diferentes reglas de asignación como "verticales" es decir entre los componentes de las reglas mismas, aminoácidos y codones. Los patrones horizontales se presentan fundamentalmente en la posición tercera de los codones y en alguna medida en la primera posición y los patrones verticales, en la primera y segunda posición. La diferente naturaleza de estos patrones y su distribución desigual de acuerdo con la posición podrían ser la impronta de la confluencia diacrónica o sincrónica de fuerzas y mecanismos evolutivos diferentes.
La Teoría coevolutiva del código genético y de las rutas biosintéticas de aminoácidos en su versión más actual trata explicar el hecho de que algunos de los aminoácidos conectados biosintéticamente tienen la misma base en la primera posición, la Teoría Estereoquímica trata de interpretar la asociación entre la naturaleza de la base que se encuentra en la segunda posición y la del aminoácido. La organización de las reglas en bloques de modo que en muchos casos los mismos aminoácidos están asignados a tripletes que se diferencian sólo en la naturaleza de la base que se encuentra en la tercera posición o aminoácidos parecidos de acuerdo a determinadas propiedades químico-físicas están asignados a tripletes vecinos con la misma base en la segunda posición codónica.Tales propiedades estructurales del código se explican dentro de la teoría de la optimización del código genético según la cual éste actúa como una suerte de filtro de errores ocurran estos en los codones durante los procesos básicos de la replicación o la transcripción o a nivel de la traducción es decir cuando se afecta la función básica de reconocimiento del significado establecido de estos tripletes.
Todas estas teorías perfectamente conciliables entre sí explican diferentes momentos y fuerzas que han contribuido a darle al código su forma actual. Este en un principio sólo contenía un subconjunto de los 22 aminoácidos codificados en los organismos actuales, presumiblemente aquel formado por los aa sintetizados en simulaciones experimentales de supuestas condiciones prebióticas, luego este se fue expandiendo en una serie de asignaciones de novo y reasignaciones a fin de incrementar la versatilidad y adaptabilidad del fenotipo, dado que los organismos primitivos contenían un aparato de corrección y decodificación mucho más rudimentario la probabilidad de errores a todos los niveles sería mayor por lo que la organización de los aa que se fueron añadiendo de acuerdo con su similitud físico-química sería una importante fuerza de minimización que contrarrestaría un poco (o por el contrario actuaría en la misma dirección) la tendencia a la asignación según la conexión biosintética y la dinámica de la interacción base nitrogenada-aminoácido.
La bioinformática ha jugado un rol importante en la validación o invalidación de todas estas teorías que intentan explicar regularidades visibles en el CG así como en la evaluación del efecto de estas características estructurales en las secuencias biológicas actuales por medio del sesgo en el uso de codones, uso de aminoácidos y tasa de substituciones sinónimas o no sinónimas. De manera general su contribución ha sido la siguiente:
-Estimación de cuán óptimo es el código genético como filtro de errores de acuerdo con determinadas propiedades mediante simulación numérica o por vía analítica. (Ver fig. 2 y 3)
- Polémica acerca de la Teoría coevolutiva en su primera versión basada en los pares de aminoácidos precursores-producto, o en su última versión basada en las familias biosintéticas de aa asociadas a la primera posición codónica resuelta esta por vía de simulaciones Montecarlo.
- Reconstrucciones de escenarios primitivos cuando surge y se desarrolla el código genético, explicación por esta vía de algunas de sus características, como el tamaño de los codones, cantidad de aminoácidos codificados, etc. en estas han jugado un rol fundamental algoritmos básicos de la bioinformática para la predicción de estructuras de ARN y de proteínas, algoritmos de alineamiento etc.
- Simulación numérica de la interacción evolutiva entre Mensaje y Código.
- Evaluación de cuan distante son los códigos más óptimos con relación al Código Genético Standard, en estos trabajos se emplean algoritmos de optimización como Simulated Annealing o algoritmos genéticos, etc.
- Cuantificar el efecto de diferentes propiedades estructurales del Código Genético Standard en el sesgo del uso de codones, uso de aminoácidos o tasa de substitución no sinónima.
- Robustez del CGS para los diferentes tipos de transiciones y transversiones.
- Simulaciones de las fuerzas selectivas o neutrales que están detrás de los cambios de reglas de asignación que han dado lugar a las variantes conocidas actualmente del código genético.
- Análisis filogenético con ARNt, Aminoacil-ARNt sintetasas o factores de liberación para determinar la ruta de determinado cambio en el CGS o la factibilidad de alguno de sus mecanismos evolutivos.
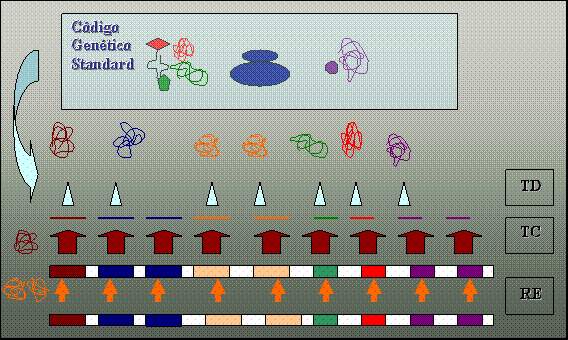
Fig 1. Esquematización de los procesos básicos de un organismo típico, TD: Traducción, TC: Transcripción, RE: Replicación. Los componentes del aparato de traducción se encuentran encuadrados en la parte superior (colores: Azul, Rojo, morado y verde). En la parte inferior izquierda se encuentran la Replicasa: Naranja y la ARN polimerasa: Carmelita oscuro. La ausencia de fidelidad de estos procesos básicos de decodificación de la información genómica sobre todo en organismos primitivos hizo particularmente importante la semantización o resemantización de tripletes vecinos con aminoácidos de propiedades parecidas (Hidrofobicidad según Kyte-Doolittle y Requerimiento polar).
1.1 Teoría estereoquímica de origen y evolución del CGS.
De acuerdo con esta teoría los patrones que se observan en el CGS se deben a la interacción entre aminoácidos y codones propios, anticodones, dobles hélices codon-anticodón, o complejo de cuatro nuecleótidos que se encuentra en el brazo aceptor, determinados nucleótidos o dinucleótidos. En una primera etapa, dada las condiciones tecnológicas más limitadas se buscaban relaciones experimentales indirectas por ejemplo el Requerimiento Polar que no es más que el coeficiente de partición de los aminoacidos en el sistema agua-piridina, este varía de acuerdo con la naturaleza de la base en la segunda posición, otros ejemplos serían la correlación entre hidrofobicidad de dinucleótidos (primera y segunda posición codónica) o de nucleótidos y aminoácidos o entre el volumen de Van der Vaals de aminoácidos y energía libre de interacción por puente de hidrógeno codón-anticodón. Todos estos casos, salvo el último, se asumían como evidencias de una interacción directa entre aminoácido y ARN.
Actualmente mediante experimentos de amplificación/selección in vitro se han aislado aptámeros de ARN con sitios de unión a varios aminoácidos como la arginina, valina, isoleucina entre otros, ricos estos en sus respectivos codones o anticodones.
En el mundo ARN se piensa que los aminoácidos unidos por interacciones covalentes o no a ribozimas primitivas, fueron en un principio cofactores, luego ribozimas que catalizaban la síntesis de pequeños péptidos fueron reemplazadas por complejos de proteínas, ARNm y ARNt muy similares al actual aparato de traducción. (Ver fig.1)
Es interesante que la interacción de un aa con un triplete ARN dado es muy similar a la que hay entre este y un triplete vecino, o la que habría entre aa parecidos con tripletes vecinos entre si, es decir la función minimizadora provendría de una ley puramente química y no del esquema: asignación aleatoria-selección natural.
1.2 Teoría de la Minimización de errores.
Cuando emergen los ARNt o los precursores de estos el movimiento adaptativo de asignaciones y reasignaciones de aminoácidos a tripletes se libera de restricciones químicas, con este paso previo es mucho más plausible el relajamiento combinatorio necesario para explorar múltiples códigos posibles y anclarse selectivamente en mínimos locales uno de los cuales sería el CGS o una de sus variantes coetáneas.
Una de las limitantes fundamentales para esta teoría fue la idea del "accidente congelado" apoyada en el hecho de que un cambio de regla de asignación se amplificaría necesariamente en una avalancha de mutaciones a nivel de todo el proteoma, terminando en un colapso, y la probabilidad de que esto ocurra sería mayor en organismos más complejos. Cuando se descubren variaciones nucleares y organulares del CGS cambia este paradigma.
Casi inmediatamente después de haberse descifrado completamente el CGS, en 1965 Sonneborn propone la teoría de la mutación letal, de acuerdo con la cual las mutaciones son frecuentemente deletéreas es por ello que el CGS tiene asignados aminoácidos con propiedades similares a tripletes que se diferencian en solo una base, sobre todo en la tercera posición y en la primera, unos años después Woese propone la teoría de los errores de traducción, en las primeras etapas la decodificación era muy inestable e ineficiente, los numerosos errores traduccionales la mayoría de los cuales implicaban la lectura de cada codon como su vecino hacían que de cada protogen se expresara un conjunto de polipeptidos, luego para refinar aún más la relación adaptativa Genotipo-fenotipo-ambiente la evolución favorece la asignación más precisa de aminoácidos parecidos a tripletes similares. Desde el trabajo inicial de Alff-Steinberger hasta la actualidad se han ido desarrollando métodos para cuantificar el grado de optimización del código genético con respecto a códigos aleatorios. Hay dos tendencias fundamentales:
- Probabilística: Esta consiste en obtener muestras aleatorias de códigos posibles y realizar un conteo de cuantos de estos tienen una desviación cuadrática media con relación al cambio de una base y para determinada propiedad aminoacídica inferior a la del código canónico. Con esta función de minimización se han explorado varios espacios fisico-químicos: Requerimiento polar, hidrofobicidad, punto isoelectrico, tamaño molecular, refractividad etc. Obteniéndose los mejores resultados con las dos primeras. A la función se le han incluido la influencia de varias variables, tasa de transversión o transición por posición codónica, sesgo en el uso de codones o uso de aminoácidos. Los esquemas de simulación numérica se han ido desarrollando con nuevas restricciones, aleatorización por bloques, total, fijando grupos de codones con la misma base en una posición codónica dada o incluso fijando también la naturaleza de esta base. (Ver Fig. 2 y 3). Desarrollo de nuevas medidas de optimización estructural basadas en códigos genéticos representados como grafos.
- Optimización computacional: Es decir en este caso mediante algoritmos de optimización clásicos se han obtenido códigos genéticos más óptimos con respecto al CGS, que luego se comparan con el código genético Standard. Se han empleado también métodos analíticos para cuantificar el grado de optimización del CGS.
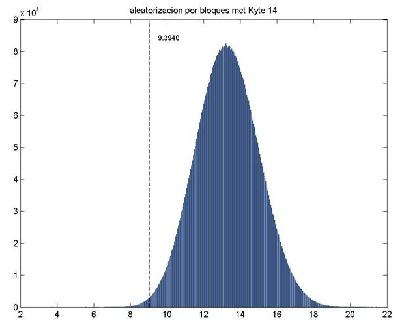
Fig 2. Monte Carlo por bloques del Código Genético Standard (CGS), histograma que representa una muestra aleatoria de 5x107códigos genéticos posibles proveniente de una población del orden 1018. La función es diferencia media cuadrática entre las magnitudes de Hidrofobicidad (Kyte,1982). La línea vertical discontinua repesenta la posición del código genético estándar.
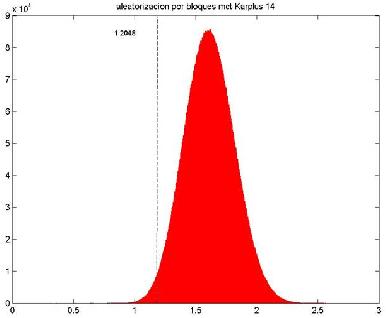
Fig. 3. Monte Carlo por bloques del Código Genético Standard (CGS), histograma que representa una muestra aleatoria de 5x107 códigos genéticos posibles proveniente de una población del orden 1018. En este caso se trata de las mismas funciones Diferencia Cuadrática Media pero con otro índice de hidrofobicidad (Karplus, 1995). La línea vertical discontinua repesenta la posición del código genético estándar.
1.3 Teoría Coevolutiva del código genético y de la biosíntesis de aminoácidos.
De acuerdo con esta teoría los aminoácidos se fueron codificando secuencialmente, según se fueran estableciendo y madurando sus respectivas rutas biosintéticas de modo que a aminoácidos conectados metabólicamente con reacciones de interconversión energéticamente favorables se le adjudicaban codones con un nucleótido de diferencia sea por ser estos parecidos químicamente, por la interacción aa-triplete, o por la interacción del nuevo aminoácido o de algún intermediario metabólico de este con el sitio de unión al aminoácido reemplazado en las Aminoacil-ARNt sintetasas. En el planteamiento inicial de esta teoría se establecían pares de aminoácidos precursor-producto y partiendo de la distribución probabilística de una medida de contiguidad de estos pares, se cuantificaba cuan alejada de la aleatoriedad estaba esta cercanía, luego se demostró la falsedad de algunos de los supuestos iniciales. En otra línea de desarrollo se ha encontrado que aminoácidos incluidos en las mismas familias biosintéticas, familia del shikimato, del aspartato o del glutamato, tienen la misma base en la primera posición codónica, los aminoácidos más primitivos están asignados a codones del tipo GNN, luego el resto se fue añadiendo a los codones CNN, ANN y UNN. Suponiendo que la función de minimización estaría restringida por este patrón de expansión del código se ha determinado mediante Monte Carlo que sus huellas prácticamente no se notan en el CGS actual. Por otra parte, se han sugerido algunas huellas de este proceso de expansión mediante el análisis filogenético de ARN y de Aminoacil ARNt sintetasas.
1.4 Efecto del CGS en las secuencias biológicas actuales.
Todos los modelos evolutivos a nivel de las secuencias de proteínas (Matrices de substitución PAM, BLOSUM etc.) contienen implícitamente la estructura del código genético, esta es bien visible sobre todo cuando se compara como varía la tasa de substitución por pares de aminoácidos para conjuntos de proteínas de diferente grado de divergencia evolutiva. Se han estudiado en las secuencias actuales el efecto de la estructura del CGS en el sesgo de uso de codones, uso de aa o tasa de substitución no sinónima. Se han propuesto modelos evolutivos que contienen explícitamente la estructura del código genético como el Modelo de Clases de Degeneración con el que se han podido hacer algunas predicciones. Otros modelos estocásticos que tratan de explicar la interacción dinámica de repercusión bidireccional entre código y mensaje con simulaciones in silico.
1.5 Variantes del CGS.Evolución y adaptabilidad.
Se han descubierto más de una treintena de variantes nucleares y organulares del Código Genético Standard, la flexibilidad evolutiva proviene de varias fuentes de mutaciones, en el ARNt, en su brazo anticodón , en el brazo aceptor o incluso en cualquiera de los otros, en las enzimas de modificación de bases del ARNt, en la Aminoacil ARNt sintetasas, en las proteínas exportación de aminoacil-ARNt o en los factores de liberación. Varios mecanismos evolutivos se han propuesto básicamente tres, dos seleccionistas y uno puramente neutralista. El último se basa en fluctuaciones drásticas en el uso de codones debido a cambios en la presión mutacional, en el caso de los dos primeros, uno consiste en la llamada teoría de la captura de codones, la cual se basa en la existencia temporal de codones polisémicos en los que se va favoreciendo uno u otro significado en virtud de los cambios en la presión evolutiva, la otra teoría explica la pérdida de determinados ARNt por las ventajas que proporciona la reducción de tamaño genómico en mitocondrias por ejemplo. En general las predicciones que se derivan de estos tres mecanismos se cumplen para diferentes organismos.
Tal vez las aplicaciones bioinformáticas más complejas sean aquellas que buscan mediante elaborados experimentos in silico recrear escenarios evolutivos que expliquen de manera congruente la aparición y cambios en los diferentes componentes del complejísimo aparato traduccional que tiene por centro el código genético. Se han propuesto explicaciones para cuestiones tan disímiles como el tamaño de los codones, de los ARNt, clases de degeneración, existencia de los ARNt, interacción coevolutiva código-mensaje, desaparición paulatina del rol que tuvo el ARN en el mundo primitivo al ser usurpada su función catalizadora por las proteínas o su función de replicación y conservación de la información por el ADN, muchos indicios de su época de esplendor aún se resisten a desaparecer en ribonucleoproteínas (ribosomas, spliceosoma o partícula de reconocimiento de la señal por ejemplo), intrones con actividad autoeditora, virus, viroides o virusoides ARN, coenzimas etc.
Bibliografía
- J Lehman J. Theor. Biol. (2000) 202: 129-144.
- R Knight, L Landweber RNA (2000) 6: 499-510.
- G Sella DH Ardell J Mol Evol (2002) 54:638-651.
- PH Von Hippel Science (1998) 281: 660-665.
- RD Knight S J Freeland L Landweber TIBS(1999),
24: 241-246.
- OP Judson D Haydon J Mol Evol (1999) 49: 539-550.
- M Archetti J Mol Evol (2004) 59:258-266.
- DA McClellan J Mol Evol (2000) 51:185-193.
- L Ribas de Pouplana, R Turner, B Steer y P.
Schimmel Pnas USA (1998) 95: 11295-11300.
- RD Knight S J Freeland L Landweber Nature (2001)
2: 49-58.
- M Ibba D Soll Science (1999), 286:1893-1897.
- R Amirnovin J Mol Evol (1997) 44:473-476.
- M Di Giulio M Medugno J Mol Evol (2000) 50:
258-263.
- S J Freeland RD Knight L Landweber y L D Hurst
Mol. Biol. Evol. (2000) 17: 511- 518.
- S J Freeland T Wu y N Keulmann Orig Life and
Evol. Biosph. 33: 457-477.
- D Gilis, S Massar, N J Cerf y M Rooman Genome
biology (2001) 2 (11).
|

